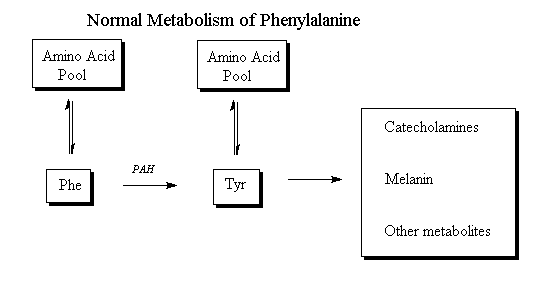
Normally, our body will breakdown phenylalanine to tyrosine which will then form catecholamine (Hormone such as dopamine formed in the adrenal gland), melanin and other hormones. This is usually the main route of phenylalanine while the minor route is the transamination to phenylpyruvate, which will be subsequently metabolized.
Hydroxylation of phenylalanine
The enzyme catalyzing the reaction is phenylalanine hydroxylase (PAH), a mixed-function mono-oxygenase that uses molecular oxygen. This enzyme also uses the cofactor tetrahydrobiopterin (BH4), which is oxidized in the course of the reaction to dihydrobiopterin (BH2). The cofactor must be regenerated by a separate system of enzymes for PAH action to continue.
Native PAH is a polymeric enzyme; it is not yet clear if the subunits are identical, but SDS-polyacrylamide electrophoresis shows two 50kDa subunit bands. However, there is only one genetic locus for PAH.
Transamination of Amino Acids
Transaminases catalyze the transfer of -NH2 groups from the amino acids, onto alpha-ketoglutarate. Many different transaminases are known, and they are generally of broad specificity for amino acids (that is, one enzyme can accept as substrates two or more different amino acids). All have the same cofactor requirement - pyridoxal phosphate (vitamin B6).
Transamination of phenylalanine to phenylpyruvate is normally of negligible importance, so long as the main route is functioning. However, if the main route is blocked for some reason, then transamination of Phe becomes quite important. In fact, the production of the distinctive minor metabolite, phenylpyruvate, can be used to diagnose deficiencies in the main route of metabolism of phenylalanine.

Abnormal Metabolism
Under hyperphenylalanine conditions, where there is too much phenylalanine in the body, the body will resort to convert the phenylalanine through the transamination pathway, converting phenylalanine to phenylpyruvate.
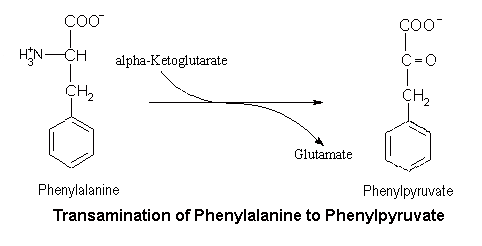
The role of alpha-ketoglutarate as the acceptor of the amino group from Phe, with consequent formation of glutamate.
The phenylpyruvate is further metabolized. Decarboxylation of phenylpyruvate gives phenylacetate, while a reduction reaction gives phenyllactate. The phenylacetate can be further conjugated with glutamine to give phenylacetyl glutamine. All of these metabolites can be detected in serum and urine by suitable clinical tests.
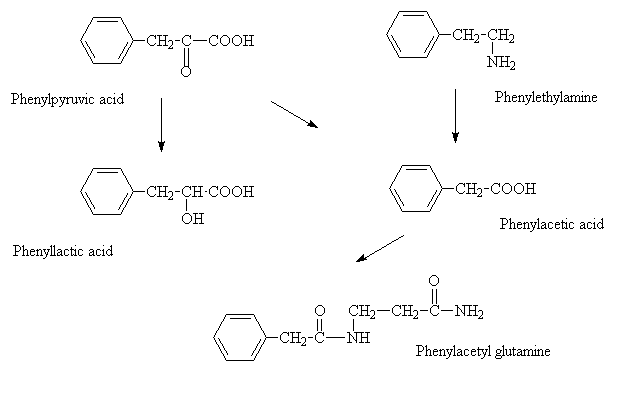
 |
| Overview of Phenylalanine Metabolic Pathway in PKU. |
For phenylalanine hydroxylase enzyme, is the molecular oxygen the coenzyme?
ReplyDeleteif a person has phenylketonuria, am i right to say that he will have lower levels of tyrosine? and if that's the case how will the body try to increase the level of tyrosine?
ReplyDeleteHi Guo Hao! :)
Delete1) Molecular oxygen is NOT the conenzyme.
2) Yes, you are right. Their body will get the necessary amino acids including tyrosine from the specially formulated amino acid food. This has to be taken throughout their entire life.