Rare genetic disorder leads Holladay family to healthy eating habits
Published: Sunday, Dec. 2 2012 5:28 p.m. MST
 |
| Seth Oliver, 3, has a banana for a snack while his mother has her pen ready to jot it down in his notebook at their Holladay home Friday, Nov. 30, 2012. |
HOLLADAY — Making sure kids eat their vegetables is hard enough for some moms, but watching, weighing and measuring every morsel of everything they eat is another story.
"I know every bite of food my kids have eaten, every day of their lives," said Amy Oliver, a Holladay mother of three and part-time attorney. Two of her kids require extra attention, but one wouldn't know it just by looking at them.
"The only way you can tell is if you watched them eat, and even then, if you watched carefully," she said.
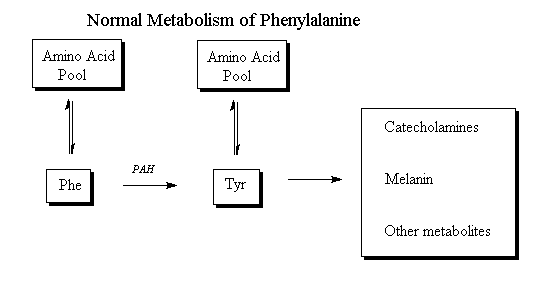
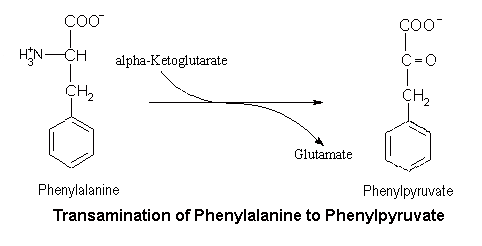
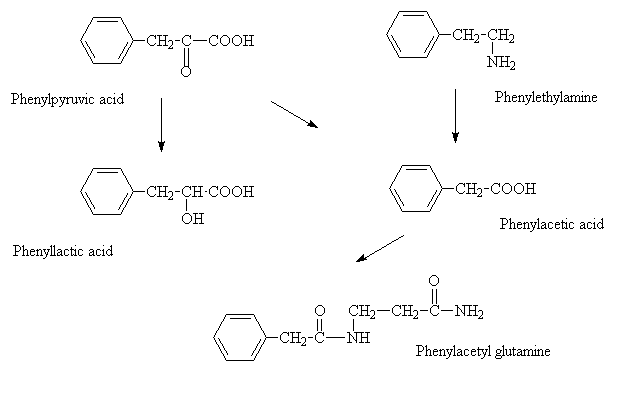
Claire, 5, and 3-year-old Seth Oliver have phenylketonuria, or PKU, a rare genetic disorder that affects a person's ability to process an important part of protein called phenylalanine. They are two of about 92 children and hundreds of adults in Utah who are diagnosed with the very rare condition, which requires scrupulous note taking on the only known treatment — a special diet for life.
Over the years, Oliver has grown accustomed to subtracting the weight of the small, color-coded plastic dishes she uses to serve the kids, the leftover peels from various foods, and whatever food her children don't eat, all the while meticulously calculating the results to get the amount of protein each child has received each day.
Claire and Seth each need about 22 grams of protein, or 1,500 milligrams of "phe" each day in order to be healthy. The amount won't change much as they get older or as they grow bigger, but it has to be monitored to prevent dangerous side effects, including irreversible brain damage and behavioural issues.
Oliver and her husband have an older son, Luke, who didn't receive the mutant gene from each of his parents and therefore can eat a normal diet.
One in 50 adults are carriers of PKU, and every person carries up to 50 defective genes that could be passed on to their children, said Dr. Nicola Longo, a professor and the chief of the division of medical genetics at the University of Utah.
There's a one in 2,500 chance that two adults who each carry the gene will come together to have a child with the metabolic disorder, and the odds are one in four for each pregnancy, he said. One in 15,000 children are born with the condition, and it is typically caught within the first week of life.
Newborn screening, in which a tiny amount of blood is drawn from the heel of a baby, checks not only for PKU, but for about 37 other metabolic disorders. The two-test series is less than $100 but critical for proper development, Longo said.
"If you find out later, the child might be dead or have irreversible mental disabilities," he said. "If you wait for symptoms, it is just too late."
Symptoms are evidenced in delayed achievement of normal developmental milestones, as the brain fails to grow. Delayed diagnosis and treatment can lead to mental retardation and other neurological problems, such as memory loss and mood disorders.
Longo said newborn screening — the first test of which is performed within 72 hours of birth, and the second between 7 and 21 days of age — has been conducted in the United States and Europe for the past 40 or more years. It was initiated in 1963 and mandated by law in the U.S. in 2008. Unless parents opt out, it is routinely conducted, saving lives and brain function, Longo said.
He said, however, there are still patients who are diagnosed later in life with PKU after mental disabilities arise because they were not diagnosed at birth. Those individuals, Longo said, are often institutionalized and cannot live normal lives.
Adults who do not follow the stringent diet can slip mentally, including loss of various executive functions. Longo said the effects are reversible if a person returns to the diet, but that is easier said than done.
"They often do not realize they are experiencing problems, and if they do not know, they cannot fix it," he said. "They end up having problems doing their jobs, with relationships, but they can still live relatively normal lives. It is just harder to do so."
The Oliver children are learning as they grow, and so far, they don't know any different.
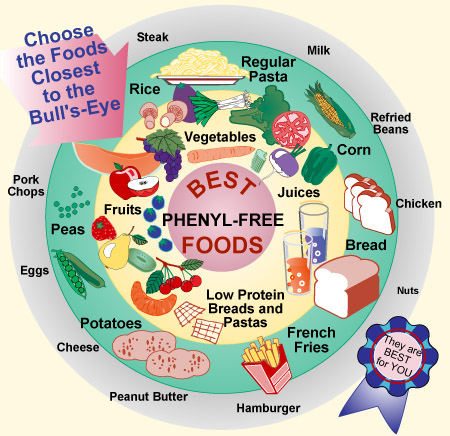
Claire knows she can only eat "low-phe" foods, and the family tries to incorporate as much of the kids' diet into their daily lives, "recognizing it is always going to be different," Oliver said.
She's lost her own taste for meat, and the whole family eats a lot healthier, she said.
Most of the children's meals have to be homemade, aside from the occasional frozen vegetables that can be warmed in the microwave.
"There are no convenience foods for us," Oliver said.
She often uses Metamucil to give bread made with rice flour added bulk and fiber. The kids are allowed minimal spices and flavorings, but they enjoy a little bit of butter on otherwise bland mashed potatoes.
To make up for the lack of protein needed for optimal growth, Claire and Seth, and other patients of PKU, have to drink special formula, a synthetic milk product that has phenylalanine removed. The formula can cost a family up to $12,000 a year, but in Utah, it's mandated to be covered on most insurances, said Oliver, who started a local support group called the Intermountain PKU and Allied Disorders Association in 2008.
"As a mother, the most reassuring thing is to see other kids with PKU running around, being normal kids," she said. "It's also nice to be able to talk to other mothers about what we go through."
In addition to raising awareness and fundraising for research, the association initiates parents of newly diagnosed PKU patients with a starter kit containing a food scale, calculator, cookbook and various other resources, which Oliver said quickly become the tools of the trade, as they go everywhere with parents of children with PKU.
"You have to be really organized," she said. "We never do anything spur of the moment, and we carry supplies with us everywhere we go."
A typical day includes measured amounts of a variety of fruits and vegetables, crackers, low-protein cheese, popcorn and no meat, dairy, eggs, beans or nuts, which are forbidden on the special diet.
"You don't want any of your children to have a health condition of something they have to deal with for the rest of their lives, but the silver lining is that they have each other in this," Oliver said.
Claire and Seth are closely monitored by geneticists at the U., who follow their growth and development, but also their dietary needs, which can change over time.
Oliver admits the diagnosis was daunting at first, but "if it's your child's life on the line, you could do anything for them," she said.
"We're just like anybody else. We just eat differently. We eat to live. We view food as a necessity," Oliver said.
A growing understanding of food issues in the general public, including gluten intolerance and various allergies, have made the family feel more at home when they go out, and explaining their sometimes awkward habits of weighing and measuring foods gets easier as they go.
"They know they can't eat anything without clearing it with us first," Oliver said, as she sliced an apple Seth picked from the large fruit basket poised on the kitchen counter.
It's nice, she said, that they want to eat healthy foods, but it is also comforting for her, knowing that they have to in order to thrive.









{kind=link}
{kind=link}
{kind=link}
{kind=link}